ENDOGENOUS SYSTEMS INVOLVED IN EXERCISE-INDUCED ANALGESIA
INTRODUCTION
For more than 30 years, studies have shown the analgesic effect produced by exercise in healthy subjects and chronic pain conditions and have expanded these studies to investigate the complex molecular mechanisms involved in exercise-induced analgesia (1, 2).
Although studies have found that exercise produces an analgesic effect, prescription remains difficult in some cases, since the optimal exercise parameters such as type and intensity are still not well defined (3, 4). Furthermore, not all types of pain conditions respond equally well to exercise-induced analgesia.
Exercise-induced analgesia has been reported during and after different types of exercise, such as aerobic, resistance, and isometric (1, 5). This effect is most prominent with higher intensity and longer duration of aerobic and isometric exercises. Few studies have investigated the different load of resistance- exercise on the nociceptive threshold. The first studies that investigated exercise-induced analgesia used mechanical and electrical stimuli to evaluate this effect. Later studies used thermal stimuli, but the results were somewhat inconsistent (3, 6, 7). Regarding the influence of age on this effect, studies using different modes of exercise have found pain and functional improvement in older adults with chronic pain (4, 8). In animals, a study investigated the effects of aging on walking-induced analgesia using a formalin-induced paw licking test. The authors found that the analgesic effect induced by walking was age-dependent with the oldest mice exhibited the least analgesia (4 > 24 > 48 weeks) (9). Studies investigating sex differences in exercise-induced analgesia have produced inconsistent results. A study showed that only women reported increase in nociceptive thresholds after two brief maximal handgrip contractions. However, when pain ratings were evaluated, both men and women reported decreased pain (10). In addition, other studies have found that isometric contractions increased the nociceptive threshold in men and women (11, 12). Comparing the sex differences in the nociceptive threshold of older individuals, a study found that older women reported greater pain sensitivity, higher pain ratings, and larger reductions in pain ratings than men after isometric exercise (5). Independent of athletic status, a study showed that treadmill running for 10 min at 85% induced reductions in ratings of pain intensity and unpleasantness during a cold pressor test in women but not in men (13).
Although exercise-induced analgesia is well documented in the literature, there are several hypotheses that attempt to unravel the cause of this effect. The most accepted of these hypotheses is the activation of endogenous systems, such as the opioidergic, nitrergic, serotoninergic, noradrenergic, and endocannabinoidergic systems and of anti-inflammatory cytokines. Additional hypotheses have been proposed, such as activation of high threshold motor units, activation of the primary motor cortex and corticospinal tracts, and interaction of the cardiovascular and pain modulating systems. In this sense, the present study aimed to determine which of the previously implicated endogenous systems are involved in exercise-induced analgesia.
ENDOGENOUS SYSTEMS INVOLVED IN EXERCISE-INDUCED ANALGESIA
Opioidergic system
The discovery of endogenous opioids started after Hughes and Kosterlitz (14), in 1974, isolated enkephalins from pig brain. Around the same time, Simantov and Snyder (15) found a similar substance in calf brain and named it ‘endorphin’, which is an abbreviation of ‘endogenous morphine’.
The endogenous opioid system includes a large number of opioid peptides, such as endorphins, enkephalins, and dynorphins, which are derived from the precursor polypeptides, pro-opiomelanocortin, proenkephalin, and prodynorphin, respectively (16). These peptides bind to opioid receptors; three primary opioid receptor types mediate analgesia and are designated µ, k, and d. The interaction between G protein-coupled opioid receptors and their effectors can be direct or involve intermediate or other indirect effector pathways (17). The activation of inward-rectifying K+ channels, the inhibition of voltage-dependent Ca2+ channels and the inhibition of adenylyl cyclase are direct effects of the activation of G protein-coupled opioid receptors (17). Conversely, these opioid receptors can also activate phospholipase C (PLC), the mitogen-activated protein kinase (MAPK) cascade, and large conductance Ca2+-activated K+ channels by utilizing intermediary messenger systems (18). Modification of the Ca2+ and K+ channel currents by opioid receptors can lead to a decrease in neuronal excitability, a decrease in the neuronal firing rate, and inhibition of neurotransmitter release (17). These receptors are largely located in all areas of the central nervous system (CNS), including areas involved in analgesia (the brainstem, medial thalamus, spinal cord, hypothalamus, and limbic system). Furthermore, opioid receptors have also been identified in the peripheral nervous system (15).
Basbaum and Fields (19) proposed that the analgesic action of endogenous results from activation of excitatory connections between the periaqueductal gray area (PAG) and the nucleus raphe magnus. Nucleus raphe magnus neurons, in turn, project, via a pathway in the dorsal part of the lateral funiculus (DLF) of the spinal cord to the region of nociceptors in the spinal dorsal horn, and its trigeminal equivalent, the nucleus caudalis. These raphe-spinal neurons selectively inhibit dorsal horn nociceptive neurons, including interneurons and a population of rostrally projecting spinothalamic and spinoreticular neurons (20).
In addition, several studies have demonstrated an increase in the plasma levels of endogenous opioids, mostly β-endorphin, during and after exercise. They found an increase in β-endorphin levels during aerobic exercise with an intensity of 85% of the maximum heart rate in women trained to exercise to exhaustion (21, 22). Furthermore, these studies have demonstrated that this increase is directly related to the intensity applied. High β-endorphin levels were found after 20 min of stationary bike exercise, only increasing to 80% of VO2Max when compared to 40% and 60% VO2Max. In addition, other studies found that β-endorphin levels change after running between 40 – 70% VO2Max (21, 22). However, another study that compared a progressive exercise protocol of 60%, 70% and 80% VO2Max for 1 hour demonstrated that each intensity similarly produced a significant increase in β-endorphin levels (23). Similar to aerobic exercise, studies also have found an increase in β-endorphin levels after resistance exercise. The first study compared burst activity resistance exercise (weight lifting) to aerobic exercise (treadmill) in five males and found a similar increase in β-endorphin levels between the exercises (24). In addition, an increase in plasma β-endorphin concentrations was found after heavy-resistance exercise in eight healthy males (25). However, similar to studies using aerobic exercise, there were contradictory results from studies performed using resistance exercise. There was no difference between the β-endorphin levels of six resistance-trained athletes who had participated in a three-set series of eight repetitions of an isotonic exercise performed at 80% maximal effort (26). Furthermore, these same authors found in another study that β-endorphin levels were reduced after three sets of eight repetitions of isotonic resistance exercise at 80% of one repetition maximum in males and females when compared to the levels before exercise (27). However, none of these previously described studies associated these increases with analgesia evaluation.
Thus, after studies demonstrated the involvement of endogenous opioids in analgesia and that exercise produces an increase in β-endorphin levels, several studies have begun to investigate the participation of endogenous opioids in exercise-induced analgesia.
In a study that evaluated 8 men and 3 women who were subjected to exercise protocols (20 min of leg and arm exercises) and transcutaneous nerve stimulation (TENS, low frequency) exercise was shown to significantly increase the dental pain threshold of the subjects, which gradually returned to baseline levels 50 min after exercise. The analgesic effect found after 20 min of arm and leg exercise was reversed by naloxone (0.8 mg, a non-selective opioid receptor antagonist) injected (i.v.) (28).
A double-blind study, performed with eight patients diagnosed with symptomatic myocardial ischemia and nine with asymptomatic myocardial ischemia was compared during physical exercise under naloxone (6 mg, i.v.) or placebo. Ischemic (a tourniquet on the forearm, under control of transcutaneous PO2) and electric nociceptive thresholds (intracutaneous electrode placed in the finger with the electrical stimulus under computer control and two-interval forced-choice psychophysical technique) and plasma β-endorphin levels were assessed before, during and following exercise. Results indicated that ischemic and electrical pain thresholds were higher in the asymptomatic patients compared to symptomatic patients at baseline. A moderate, but statistically insignificant, increase in pain threshold was found following exercise. Naloxone i.v. administration attenuated ischemic pain threshold following exercise but had no effect on electrical pain threshold. Plasma β-endorphin levels were found to increase during exercise, with significantly higher increases in the asymptomatic patients compared to symptomatic patients. In addition, naloxone administration also attenuated the β-endorphin increase observed in the asymptomatic patients (29).
A study that investigated the influence of exercise on the nociceptive threshold using multiple pain stimuli showed that cold pressor pain did not influence 12 runners after 10 km run at 85% of maximal aerobic capacity; however, the thermal and ischemic pain were attenuated in the same participants following exercise and this effect was associated with increase of β-endorphin plasma levels. In addition, it was found that i.v. injection of naloxone (8 mg) reversed the post-run analgesic response for ischemic stimulation but not for thermal stimulation (30). Furthermore, sensory-decision theory analysis was employed to assess both the discriminability of pain stimuli and pain report criterion of three different stimuli (thermal, ischemic and cold pressor). The discriminability represents how well a person can distinguish between different intensities of painful stimuli, while the pain report criterion refers to the person’s willingness to report a stimulus as painful. The results revealed that thermal discriminability and ischemic discriminability were significantly reduced following running, indicating that an analgesic response had occurred (30).
In addition, besides pain stimuli, the antagonist dose also influenced the analgesic response found after exercise. A study demonstrated that treatment with 2 mg naloxone completely blocked the analgesic effect, caused by a 1-mile run, on pressure pain thresholds in 9 men and 6 women when compared to a 10 mg dose (31). However, another study demonstrated that high-dose naloxone (20 mg) did not influence the analgesic response, evaluated by the response to electrical stimuli applied to the dental pulp or pressure tests applied to the finger, in ten healthy active men that underwent exhaustive exercise on an ergometric cycle (32).
The most compelling evidence to support the theory of exercise-induced analgesia and the involvement of the endogenous opioid system have been provided by animal experimentation. Most of these studies investigated whether exercise-induced analgesia is mediated by endogenous opioid mechanisms using swimming and running as primary exercise stimuli.
The pioneering study investigating the systemic involvement of endogenous opioid systems in exercise-induced analgesia showed that different doses of naloxone (0, 1, 5, 10 and 20 mg/kg, subcutaneously (s.c.)) administered at weekly intervals produced a dose-dependent reduction of up to a maximal reduction of 50% at 20 mg/kg in the swim-induced analgesia in rats, measured by flinch-jump thresholds (33). In addition, another study evaluated the influence of various cold-water (2°C) swim parameters on exercise-induced analgesia. Male rats were exposed to (i) various durations of cold-water swims, (ii) intermittent versus continuous cold-water swims, and (iii) 60 consecutive cold-water swims in naltrexone (an opioid receptor antagonist) and saline conditions. Naltrexone subcutaneously administered 10 min before swimming partially antagonized continuous cold-water swim-induced analgesia, but at only high doses of naltrexone (21 mg/kg). However, at lower doses (14 mg/kg), naltrexone significantly antagonized intermittent cold-water swims and enhanced the analgesic response produced by 60 consecutive swims. These results demonstrated that naltrexone differentially influences cold-water swim-induced analgesia depending upon specific parameters of the exercise condition, including the duration of the swim, whether the swim was intermittent or continuous, or whether a large number of consecutive cold-water swims were completed (34). However, another study demonstrated that the swim-induced analgesia in female mice, evaluated by the response to a hot plate test, was blocked by low-dose (100 µg/kg, s.c.) naloxone administered 1 hour prior to swimming. Furthermore, these authors found that the analgesic effect produced by exercise was greater than the analgesia obtained upon injection of 15 mg/kg of morphine (35). In addition to the dose, the pre-administration time and the type of antagonist were also shown to be important factors in cold-water swim-induced analgesia. Naloxone had a more rapid effect, onset and shorter half-life than naltrexone.
The duration of exercise can also influence the analgesic response. A study compared a female mouse group that swam for 15 s with another group that swam 3 min. Analgesia was induced in both groups; however, when parallel groups were pretreated with naloxone (5 mg/kg, s.c.), analgesia was blocked in only the 3-min-swim group (36). These studies suggested that the swim duration influences the nature of analgesia and that there appear to be multiple analgesic systems (opioid and non-opioid) involved in this type of analgesia.
Studies investigating endogenous mechanisms involved resistance exercise-induced analgesia in animals is limited. Only one study using a weightlifting exercise model demonstrated the participation of the endogenous opioid system in the analgesic response. In this study, the animals underwent an acute resistance exercise protocol (15 sets of 15 repetitions) and training protocol (3 sets of 10 repetitions, three times per week for 12 weeks); in both groups, the exercise intensity began at 75% maximal repetition. The nociceptive threshold was measured by a paw-withdrawal test, which demonstrated that analgesia occurred only after acute resistance exercise, and this effect was reversed by naloxone (2 mg/kg) preadministration (37).
Regarding isometric exercise, although some studies have revealed that it induces reduction in pain ratings and increase in pain threshold (10, 39, 40), only one study investigated opioid endogenous levels after isometric exercise. This study did not find alterations in beta-endorphin levels in six elite kayak paddlers and 14 untrained subjects after an isometric knee-extension exercise (41). Furthermore, these results were not associated with nociceptive threshold evaluation.
An important study found a longer tail-flick latency in female mice that swam for 3 min in 32°C water compared to non-swimming controls, and this result correlated with a reduction of the [3H] leu-enkephalin (LE) binding in the brain homogenates of exercise-induced mice. The authors suggest that the reduced LE binding may be due to the occupation of a proportion of the opioid receptor population by an endogenous ligand, which is consistent with the interpretation of the involvement of endogenous opioids in the observed increased in tail-flick latency (42). However, no antagonist was used and no additional experiments were performed to confirm this hypothesis.
Exercise-induced analgesia was also found to be the same in both disease and pain animal models. Male spontaneously hypertensive and normotensive rats were trained to spontaneously run in running wheels. After 3 to 4 weeks of training, the pain sensitivity, measured by a squeak threshold to electrical stimulation, was reduced in both rats. Furthermore, naloxone intraperitoneal administration decreased squeak thresholds to baseline levels, indicating the involvement of the endogenous opioid system (43).
Low-intensity treadmill exercise for 5 consecutive days reduced chronic muscle pain induced by acidic saline solution in rats. This analgesic effect, evaluated by the response to von Frey filaments, was attenuated by naloxone intraperitoneal preadministration, demonstrating that the endogenous opioid system also participates in exercise-induced analgesia in the chronic muscle pain model (44). In addition, another study demonstrated that regular moderate aerobic exercise reversed neuropathy-induced thermal and tactile hypersensitivity and increased endogenous opioid (β-endorphin and met-enkephalin) content in the PAG and in the rostral ventromedial medulla (RVM); these regions are important in descending pain modulation. Furthermore, the analgesic effect induced by moderate aerobic exercise was reversed by naloxone and naltrexone, systemically or intracerebroventricularly administered, suggesting a peripheral and central involvement of opioid receptors (45).
In addition, another study demonstrated that the naloxone pretreatment (injected in the right tibiofemoral articular space) reversed the analgesia in Wistar rats with acute knee synovitis that were subjected to jumping-in-water exercise with a 50% overload (46). Opioid receptors located in peripheral nervous terminals may be activated by exogenous and endogenous opioids expressed in immune cells to produce significant analgesia (47).
The release of endogenous opioids by the adrenal gland may be another explanation for exercise-induced analgesia. A study demonstrated that analgesia was induced by a 5-day swimming period in male mice subjected to acetic acid-induced abdominal writhing but was not induced in mice with bilateral adrenalectomy or naloxone pretreatment (48).
As most studies have used naloxone or naltrexone, which are non-selective opioid receptor antagonists, it is difficult to locate a specific opioid receptor involved in exercise-induced analgesia. In addition, only one study investigated the involvement of opioid receptors in exercise-induced analgesia at the central level (45). In most studies, especially with humans, naloxone was administered systematically and in some studies, it was discovered that beyond the reversal of analgesia, a reversal of euphoria also occurred, which suggests that its effect was systemic.
Despite being one of the first mechanisms described in exercise-induced analgesia, several studies were unable to elucidate the role of endogenous opioids in this effect, which suggested the involvement of other endogenous mechanisms.
Nitrergic System
Another important endogenous candidate for exercise-related analgesia is the nitric oxide (NO) system. Some studies have demonstrated its analgesic effect and an increase in the levels of nitrate in the plasma after exercise (49, 50). NO is a soluble gas continuously synthesized in mammalian cells as a by-product of the conversion of its physiological precursor, the amino acid L-arginine, to L-citrulline. This reaction is catalyzed by one of three isoforms of enzymes known as NO synthase (NOS). Two of the NOS enzymes, specifically endothelial NOS (eNOS) and neuronal NOS (nNOS), are calcium-dependent and constitutively produced at relatively low levels of NO. The inducible NOS isoform (iNOS) is expressed for a longer period of time upon activation by a variety of factors, including exercise (51). Once synthesized, NO can diffuse within the same cell or neighboring cells, where it binds to the heme group of soluble guanylyl cyclase to generate CGMP from GTP (52). A study showed that activated CGMP promotes the opening of KATP channels, which may result in analgesia (53). Thus, by intracellular mechanisms, the analgesic effect of NO is produced by the NO/cGMP/KATP pathway.
Several factors are responsible for the increase of NO production during resistance exercise. In rodent skeletal muscle, nNOS, eNOS and inductive NOS isoforms are highly expressed in muscle fibers and are activated by exercise. The increase of shear stress by increased blood flow and muscle contraction induced distortion of resistance vessels, stimulates eNOS and nNOS (54). In addition, microdamage to myofibrils during muscle contractions releases/stimulates inflammatory cells, which activate iNOS. Red blood cells release ATP in low oxygen environments and in response to deformation due to muscle contractions. ATP binds to purinergic receptors on the endothelium, leading to eNOS activation and NO production (55). Furthermore, during exercise, there is an increase in NO levels, which has been implicated in metabolic control via effects on blood delivery, glucose uptake, oxidative phosphorylation, contractility, and excitation-contraction coupling of the skeletal muscle (56).
Additionally, an increase in blood NO levels was correlated with the reduction of migraine pain severity, frequency and duration in women that underwent a regular long-term aerobic exercise protocol that consisted of exercise periods of 1 hour/day, 3 days/week for eight weeks (57). Furthermore, NO has been demonstrated to have the ability to reduce the nociceptive activity of the peripheral and central nervous system after exercise. Studies found the involvement of NO/cGMP/KATP in the analgesia produced by acute aerobic resistance exercise in rats (58, 59). In these studies, the acute aerobic exercise protocol involved the animals running on a treadmill for an average time of 59 min at a progressive velocity until the onset of fatigue, and the resistance exercise protocol involved a weight-lifting exercise model that consisted of the animals performing 15 sets of 15 repetitions with a load at 65% to 75% of 1 repetition maximum. Immediately after exercises, the mechanical and thermal nociceptive threshold was increased, which was measured by paw-withdrawal and tail-flick tests. This effect was reversed by specific inhibitors of NOS isoforms (aminoguanidine an iNOS inhibitor; L-NIO, an eNOS inhibitor and L-NPA, a nNOS inhibitor) as well as peripheral (s.c.) and central: (i.t. and i.c.v.) preadministration of cGMP and KATP (ODQ and glybenclamide). Furthermore, plasma and cerebrospinal fluid (CSF) nitrite levels were increased after exercise, and the expression of NO were increased in the dorsolateral and ventrolateral PAG. These authors suggest that dorsolateral and ventrolateral PAG contain a column of NOS-containing cells, which during exercise may release NO that can participate in the inhibitory modulation of pain (60). However, another study found that the pretreatment of unspecific inhibitors of NOS (L-NAME and L-NA, intraperitoneally (i.p.)) potentiated the swim-induced analgesia measured by tail-flick and jump tests in rats (61). Studies demonstrated that nitric oxide synthase inhibitors, such as L-NAME, may act as partial agonists stimulating instead of inhibiting NOS and guanylyl cyclase, which may contribute to the observed dual effect of NO in the nociceptive system (62, 63).
NO may have a dual role in the regulation of pain processes, i.e., it can mediate nociception or induce an antinociceptive effect. This dual effect occurs in both the central and peripheral nervous systems (64). Many studies have focused on understanding the dual effect (pro or antinociceptive) of NO, and these data indicate that this effect is dependent of NO levels and the phase of the nociceptive process (64). L-arginine, a biological precursor of NO, at a 0.1 – 1 ug dose i.pl. per paw, enhanced second-phase formalin-induced behavioral nociception, whereas at 3 ug per paw, it did not have significant effects and conversely, at 10 ug per paw, it produced antinociception resulting in a bell-shaped dose-response curve. In addition, L-NAME (a L-arginine inhibitor) i.pl., at 1 ug per paw, produced antinociception in this phase and considerably reduced the increase in second-phase nociception elicited by low doses of L-arginine (65).
Additionally, another study showed that a low dose of intrathecal administration of NO donors produced antinociception, while higher doses enhanced (i.e., caused a pronociceptive effect) or had no effect on the mechanical allodynia evoked by chronic ligature of the sciatic nerve in rats. The authors suggested that NO produces a depression in the calcium current in the same areas of nociceptive impulse conduction. However, the increase in NO concentration may in turn increase the amplitude of calcium currents, facilitating nociception (66). Rocha et al. (67) suggested that endogenous NO is an important mediator involved in the development of zymosan-induced arthritis, whereas the pharmacological administration of NO can inhibit the ongoing nociceptive phenomena. These authors demonstrated that the inhibitors of NOS decrease articular inflammatory pain induced by zimosan; however, this inhibitory effect was observed only when the inhibitors were administered before the induction of arthritis. Furthermore, it was observed that administration of NO donors induced an analgesic effect, and this effect was observed for the drugs administered after the injection of zymosan. Although these studies have revealed the dual role of nitric oxide on the nociceptive system, the studies on exercise previously cited have found that only this substance induces analgesia.
Serotonergic system
In the 1930s, Erspamer began to study the distribution of enterochromaffin cells, which stained with a reagent for indoles. The highest concentrations were found in the gastrointestinal mucosa, followed by the platelets and the CNS (68). Hence the unknown indole was named entramine. After that, Page and his colleagues at Cleveland clinic isolated and characterized a vasoconstrictor substance released from clotting blood, which was called serotonin (68). Rapport deducted that the active moiety was 5-hydroxy tryptamine, which was isolated as serotonin by Page. In 1952, Erspamer and Asero identified entramine as 5-HT. 5-HT is autacoids as well as important neurotransmitter in CNS and peripheral nervous system (PNS) (68). The serotonergic system is known to modulate pain, mood, emotion, sleep and so it is implicated in the control of numerous behavioral and physiological functions. 5-HT exerts its multiplicity of physiological effects through an unsurpassed diversity of receptors (69).
The involvement of 5-HT in the descending control of pain has long been recognized (70). The pharmacological evidence showed that electrical stimulation of the nucleus raphe magnus (NRM) increases the release of metabolites of 5-HT in the medullary dorsal horn cells in the rat spinal cord (71). Studies demonstrated that electrical or chemical stimulation of the NRM and the surrounding nucleus reticularis paragigantocellularis produced analgesia, which was antagonized by intrathecally administered 5-HT receptor blockers and was accompanied by the efflux of 5-HT from the spinal cord (72). 5-HT is generally thought to be an inhibitory neurotransmitter in the dorsal horn cell. It is involved not only in modulating pain transmission signals in the dorsal horn but is also released from this site. However, a study found that 5-HT3 receptors are involved in hyperalgesia and tolerance induced by morphine. In that study, the authors demonstrated that systemic or intrathecal injection of a 5-HT3 antagonist significantly prevented or reversed these symptoms (73). In addition, this group had previously demonstrated that morphine is strongly regulated by the expression of the Htr3a gene in various central nervous system regions including the amygdala, dorsal raphe, and periaqueductal gray matter, which have been linked to opioid dependence (74).
5-HT has been demonstrated to be involved in exercise-induced analgesia by several studies evaluating humans and animals. Studies found that analgesia induced by intermittent cold-water swims (18 pairs of 10-s swims and 10-s recovery periods at 2°C) in rats, was reduced by both systemic and intrathecal 5-HT2 antagonist receptor (methysergide, 5 – 10 mg) administration (75). Additionally, another study also demonstrated the involvement of serotonin in the analgesia induced by high-intensity extended swimming exercise in mice, which was reversed by pretreatment with r-chlorophenylalanine methyl ester (PCPA, an inhibitor of serotonin synthesis, 100 mg/kg, i.p.) (48). These results suggested that 5-HT2 participates in exercise-induced analgesia at the peripheral and spinal levels.
Exercise produces an increase in (5-HT) concentrations in most brain areas, including areas involved in pain control (76). In addition, a study demonstrated that treadmill running exercise results in the release of 5-HT in layers II, III, IV, V of the dorsal horn (77). Furthermore, rats with sciatic nerve transection that trained on a treadmill presented a higher serotoninergic immunoreactivity in the neuronal cytoplasm of NRM, the area also involved in the control of pain, which was associated with an increase in the mechanical nociceptive threshold (78). Although, these areas (dorsal horn and NRM) are importantly involved in the descending control of pain, the nociceptive threshold was not evaluated.
Exercises have been the primary mode of treatment for low back disorders, aiming to achieve pain reduction. Studies showed that spinal stabilization and treadmill walking exercises produced a significant increase in plasma 5-HT levels in participants with chronic low back pain (79, 80). However, the increase in 5-HT levels after exercise was not associated with the pain response of the participants in any of these studies..
However, in women with fibromyalgia, a study found that aerobic walking exercise three times a week for 20 weeks significantly increased 5-HT levels and its main metabolite 5-hydroxindolacetic acid (5HIAA) when compared with baseline levels; there were no differences in the pain visual analog scale values (81).
Noradrenergic system
The noradrenergic system is an essential system that becomes activated during exercise to exert important functions, such as cardiovascular control, fuel mobilization and release of hormones and neurotransmitters (i.e., catecholamines) (82). Furthermore, the noradrenergic system may also participate in exercise-induced analgesia. Catecholamines may modulate the nociceptive pathway by activation of α2-adrenergic receptors (a2-ARs) (83). The α2-ARs receptors are divided into 3 subtypes α2A, α2B, and α2C. The α2A and α2C-ARs are Gi/Go protein coupled, which inhibits cyclic adenosine monophosphate production; the resulting opening of K+ channels and closing of voltage-gated Ca2+ channels, in turn, results in hyperpolarization and a reduced rate of firing of excitable cells (84). α2-ARs have also been found in different areas involved in pain control, for example, the periaqueductal gray, Locus coeruleus and dorsal root ganglion (DRG) (85).
Few studies have investigated the involvement of the noradrenergic system in exercise-induced analgesia, all of which were performed using rodents. The first study that evaluated the participation of this system was conducted in 1985, and it examined whether acute or chronic desipramine (a selective inhibitor of the norepinephrine transporter) intraperitoneal injection could potentiate cold-water swim-induced analgesia in mice. However, the authors found that neither acute nor chronic desipramine treatment altered the analgesic effect induced by swimming (86).
Other studies using different antagonist pretreatments also demonstrated the involvement of the noradrenergic system. A study found that idazoxan, prazosin and yohimbine (α1 and α2-adrenergic antagonists) systemic and i.c.v. preadministration attenuated warm-water swim-induced analgesia in mice. Furthermore, the warm-water swim-induced analgesia was potentiated by clonidine (α2-adrenergic receptor agonist) and noradrenaline intracerebroventricular preadministration (87). Thus, this work suggests involvement of the peripheral and central levels of the noradrenergic system, via α1 and α2-adrenergic receptors in this effect. In addition, other studies have found that clonidine analgesia was potentiated by swimming, reinforcing the participation of α2-adrenergic receptors (88, 89).
The noradrenergic system was also found to be involved in the analgesia induced after running on a treadmill and resistance exercise in rats. A study demonstrated that this analgesic effect was reversed by α2, α2A, and α2C-adrenergic receptors antagonist (yohimbine, rauwolscine and BRL 44408) subcutaneous preadministration and by guanethidine (a selective inhibitor of transmission in adrenergic nerves) pretreatment. Furthermore, the analgesic effect produced by running was not observed in α2-AR knockout mice. In addition, when yohimbine was administered intrathecally or intracerebroventricularly, it did not alter antinociception induced by aerobic and resistance exercise protocols; α2-ARs expression in the rat brain did not change after both exercise protocols, revealing peripheral involvement of the noradrenergic system (90).
Endocannabinoid system
The endocannabinoid system is comprised of two G protein-coupled membrane receptors (cannabinoid CB1 and CB2 receptors) that are negatively coupled to adenylate cyclase and positively coupled to mitogen-activated protein kinase (91). These receptors have been found in the CNS, including structures that participate in the descending control of pain, such as the PAG, RVM and the dorsal horn of the spinal cord, peripheral nervous system in DRG and immune cells (91, 92). Both receptors, when activated by their endogenous ligands (endocannabinoids), such as anandamide (AEA) and 2 arachidonoylglycerol (2-AG), which are often accompanied in tissues by non-cannabinoid receptor congeners, such as palmitoylethanolamide (PEA) and oleoylethanolamide (OEA), promote hyperpolarization and a reduction in the rate of firing of excitable cells, suppressing neurotransmitter release, and a reduction in the nociceptive impulse (93). The endocannabinoid AEA is hydrolyzed by the enzyme fatty-acid amide hydrolase (FAAH), whereas the other endogenous cannabinoid receptor ligand, 2-AG, is degraded by the enzyme monoacylglycerol lipase (MGL) (93). FAAH also partly regulates the levels of PEA and OEA.
Studies have demonstrated the participation of the endocannabinoid system in important responses during exercise such as muscle vasodilatation; euphoria and bronchodilation (94). The endocannabinoid system may also be involved in movement control since cannabinoid CB1 and CB2 receptors were found in the basal ganglia and cerebellum (91, 95). Cannabinoid receptors also have been found in the hypothalamus and may participate in the control of thermoregulation during exercise (96). Furthermore, another study suggested the involvement of this system in the aerobic capacity, after demonstrating that pretreatment with Rimonabant (a cannabinoid CB1 receptor antagonist) reduced the running speed and distance in female high runner lines (97). In addition, the same studies found an increase in endocannabinoids after exercise. In 2013, a study in men provided the first evidence that the endocannabinoid system was activated by exercise, as evidenced by significant elevations in circulating plasma AEA levels but not in 2-AG levels following acute cycling or running (94). Then, another study investigated the influence of intense exercise (60 min at 55% followed by 30 min at 75% Wmax), with 11 trained male cyclists, on endocannabinoid plasma levels. The authors found significant elevations in AEA levels as well as in PEA and OEA levels during exercise and 15 min recovery, whereas 2-AG concentrations remained stable (98).
In addition, endocannabinoid responses to different intensities of exercise were examined, and significant elevations in AEA levels following moderate-intensity treadmill exercise were found but not at lower or higher intensities of exercise (99). In an animal study, Hill et al. (100) found that eight days of free access to running wheels increased the tissue content of AEA in the hippocampus. All of these previous studies indicated significant elevations in endocannabinoids levels following exercise.
However, only a limited number of studies have been conducted examining the involvement of endocannabinoid system in exercise-induced analgesia.
A study performed with fifty-eight participants who participated in submaximal isometric exercise demonstrated that a reduction in the pressure-pain threshold after exercise was associated with an increase in AEA, 2-AG, PEA and OEA plasma levels (101). In another study by the same group, which used a similar methodology, the authors found that naltrexone prevented the increase of AEA and OEA plasma levels after isometric exercise, whereas the increase of 2-AG and 2-OG plasma levels was not prevented. The authors suggested that 2-AG and 2-OG could contribute to nonopioid exercise-induced analgesia and that the opioid system may be involved in the increase of AEA and OEA following exercise (102).
In addition, studies with rats have shown that the analgesic effect induced by running and resistance exercise was reversed by cannabinoid receptor antagonists and potentiated by endocannabinoid metabolizing enzyme and anandamide reuptake inhibitors, when they were pre-administered systemically (s.c.) and centrally (i.t. and i.c.v.) (103, 104). Furthermore, these studies found an increase in cannabinoid CB1 receptor expression levels in the brain (PAG) and in the endocannabinoid plasma levels after both exercises. This indicates that acute running and resistance exercise affect the endocannabinoid system, which may participate in exercise-induced analgesia at the peripheral and central levels (103, 104). A study with mice also revealed the involvement of peripheral cannabinoid CB1 and CB2 receptors in the antinociception induced by acute long-distance running (105). In addition, another study with mice found that AMP-activated protein kinase is an intermediate effector in endocannabinoid-mediated exercise-induced by antinociception (106). This work showed that hyperalgesia induced by the formalin test was reduced after exercise only in wildtype mice, while exercise had no effect on nociception in AMPKα2 knockout mice. Furthermore, the serum levels of AEA were increased after exercise in both wildtype and AMPKα2 knockout mice, in association with decreased expression of FAAH and increased expression of the cannabinoid CB1 receptor in the spinal cord. Additionally, treatment with the cannabinoid CB1 receptor inverse agonist (AM251) prior to treadmill running reversed exercise-induced antinociception in wildtype mice. However, the combination of AM251 with AICAR (AMPK activator) restored the analgesic effect induced by exercise, indicating that AMPK affects exercise-induced antinociception downstream of endocannabinoids (106).
Anti-inflammatory cytokines
Anti-inflammatory cytokines are immunoregulatory molecules that control the proinflammatory and nociceptive responses. Major anti-inflammatory cytokines include the interleukin (IL)-1 receptor antagonist, IL-4, IL-6, IL-10, IL-11, and IL-13. These cytokines act in concert with specific cytokine inhibitors and soluble cytokine receptors to regulate the human immune response. Exercise reduced brain inflammation and this effect was associated with increase in IL-10 and reduction in proinflammatory cytokines IL-1β and tumor necrosis factor-α (TNF-α) (107).
In addition to endogenous analgesic substances, recent studies have gained attention by revealing the participation of anti-inflammatory cytokines in the mechanism of exercise-induced analgesia. A study found that regular physical activity altered the macrophage phenotype to increase IL-10 and prevent chronic muscle pain in mice. In addition, the authors demonstrated that the blockade of IL-10 systemically or locally prevented the analgesia in physically active mice, suggesting that regular physical activity increases the percentage of regulatory macrophages (M2, which secrete anti-inflammatory cytokines) in muscle tissue and that IL-10 is an essential mediator in the analgesia produced by regular physical activity (108). An earlier study had already found that exercise training inhibits inflammation in adipose tissue via both suppression of macrophage infiltration and acceleration of phenotypic switching from M1 to M2 macrophages in obese mice. However, proinflammatory cytokine release was not investigated (109).
Besides IL-10, IL-4 levels were increased in the spinal cord of mice with peripheral nerve injury exercised for 2 weeks. These levels were associated with increase in M2 and reduction in brain-derived neurotrophic factor, β-nerve growth factor, and glial cell activation (110), which are associated with sensitization of the nociceptive response.
Glia cells are involved in nociception genesis, especially in chronic conditions (111). This cell population is mainly composed of astrocytes and microglia (112), which when activated are responsible for the release of pro-inflammatory cytokines that play a relevant role in the transmission of nociceptive impulses (113). Gong et al. (114) evaluated the role of microglia after infant peripheral nerve injury and the effect of exercise on the delayed-onset of neuropathic pain in rat pups. They found that exercise shifted spinal cord microglia polarization to the M2 phenotype and reduced neuropathic pain. In addition, IL-10 increased and TNF-α (an anti-inflammatory cytokine) decreased after exercise. The intrathecal injection of the IL-10 antibody reduced exercise-induced analgesia. Thus, the exercise was effective in the treatment of delayed adolescent neuropathic pain via the modulation of microglial polarization.
Others substances may also be associated to exercise-induced analgesia. Studies have demonstrated that exercise induces increase of brain-derived neurotrophic factor (BDNF) (115,116). Although this protein is associated with central hypoxemia and fatigue induced by exercise (115, 116), evidences may support its involvement in the analgesic effect produced by exercise. A study demonstrated that midbrain infusion of BDNF decreased the behavioral paw flinch response to subcutaneous formalin injection and this effect was accompanied by an augmentation in serotonergic activity within the brain and spinal cord of rats (117). In addition, the analgesic effect produced by BDNF was reversed by naloxone preadministration (117). Furthermore, when injected within the PAG, dorsal raphe and spinal cord, BDNF increased the β-endorphin levels (118). Thus, these studies suggesting a modulatory effect of BDNF on the endogenous substances involved in the modulation of nociceptive information and supports the hypothesis of the involvement of this protein in the exercise-induced analgesia.
Another substance that may also participate of exercise-induced analgesia is irisin. Irisin has been described as a peptide hormone derived from skeletal muscles and cleaved from fibronectin type III domain containing 5 (FNDC5) in response to an induction by peroxisome proliferator-activated receptor γ (PPARγ coactivator 1α (PGC1-α) caused by exercise (119). In addition, as FNDC5 have been shown to induce the expression BDNF in the mice hippocampus in an exercise-dependent manner and irisin also is cleaved FNDC5 (120), we suggesting that this peptide may be involved in the exercise-induced analgesia. Although, a study did not find an association between exercise-induced myokine irisin and regulation of depression, anxiety and stress in obese women (121).
Several endogenous systems are involved in the analgesic effect induced by exercise. This involvement has been found under different intensities, durations, frequencies and modalities of exercise. Furthermore, despite several studies evaluating the participation of one or two endogenous systems in each exercise model used, evidence suggested that these systems may be synergistically activated and that this may occur during exercise (Fig. 1). A study showed that the analgesia produced by endogenous opioids may be achieved through the NO/CGMP pathway (122). Furthermore, NO promotes the release of serotonin, an important neurotransmitter involved in the inhibition of nociceptive impulses in the dorsal horn of the spinal cord (123). In addition, a study demonstrated that cannabinoid CB1 and CB2 receptors agonists reduced mechanical allodynia and thermal hyperalgesia by NO/GMPc/K+ATP pathway activation (124, 125). The NO/cGMP pathway also may be activated by α2-ARs. This activation was demonstrated in a study that found that the analgesia induced by xylazine, a α2-AR agonist, was reversed by NOARG (NOS inhibitor) preadministration and ODQ (cGMP inhibitor) (126). The endocannabinoid system also may activate noradrenergic neurons. This evidence was demonstrated by pretreatment with yohimbine, a α-adrenergic antagonist, which inhibited the analgesic effect induced by delta-9-tetrahydrocannabinol (Δ9-THC), the principal psychoactive constituent of Cannabis (127).
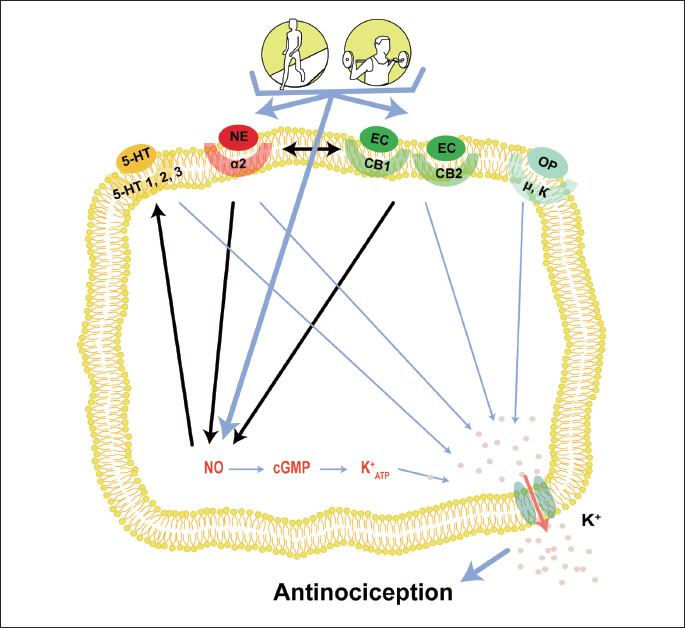
CONCLUSIONS AND FUTURE DIRECTIONS
In summary, the present review showed that a range of substances may participate in exercise-induced analgesia. Furthermore, future studies are necessary to help unravel other possible endogenous systems involved in exercise-induced analgesia and further elucidate its effects. Furthermore, knowing that exercise releases analgesic substances, investigating the intensity and modality of exercise and at what fitness level analgesia occurs may help devise effective strategies for the use of physical exercise in the treatment of different types of pain, which could ultimately lead to reduced need for pharmacological treatment.
Abbreviations: 2-AG, 2 arachidonoylglycerol; 5HIAA, metabolite 5-hydroxindolacetic acid; α2-ARs, α2-adrenergic receptors; AEA, anandamide; CNS, central nervous system; DLF, dorsal part of the lateral funiculus; DRG, dorsal root ganglion; eNOS, endothelial nitric oxide synthase; FAAH, enzyme fatty-acid amide hydrolase; HIIT, high-intensity interval training; i.c.v., intracerebroventricular; iNOS, inducible nitric oxide synthase; i.p., intraperitoneal; i.pl., intraplantar; i.t., intrathecal; i.v., intravenous; LE, leu-enkephalin; MAPK, mitogen-activated protein kinase; MGL, monoacylglycerol lipase; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; NOS, nitric oxide synthase; NRM, nucleus raphe magnus; OEA, oleoylethanolamide; PAG, periaqueductal gray matter; PCPA, r-chlorophenylalanine methyl ester; PEA, palmitoylethanolamide; PLC, phospholipase C; PNS, peripheral nervous system; RVM, rostral ventromedial medulla; s.c., subcutaneous; TENS, transcutaneous nerve stimulation; VO2Max, maximal oxygen uptake;
Acknowledgments: This work was supported by Fundacao de Amparo a Pesquisa do estado de Minas Gerais (FAPEMIG).
Conflicts of interest: None declared.
REFERENCES
- Koltyn KF. Analgesia following exercise: a review. Sports Med 2000; 29: 85-98.
- Sluka KA, O’Donnell JM, Danielson J, Rasmussen LA. Regular physical activity prevents development of chronic pain and activation of central neurons. J Appl Physiol 2013; 114: 725-733.
- Hoeger Bement MK, Rasiarmos RL, DiCapo JMT, et al. The role of the menstrual cycle phase in pain perception before and after an isometric fatiguing contraction. Eur J Appl Physiol 2009; 106: 105-112.
- Koltyn KF. Exercise-induced hypoalgesia and intensity of exercise. Sports Med 2002; 32: 477-487.
- Lemley KJ, Drewek B, Hunter SK, Hoeger Bement MK. Pain relief after isometric exercise is not task-dependent in older men and women. Med Sci Sports Exerc 2014; 46: 185-191.
- Naugle KM, Fillingim RB, Riley JL. A meta-analytic review of the hypoalgesic effects of exercise. J Pain 2012; 13: 1139-1150.
- Cook DB, Koltyn KF. Pain and exercise. Int J Sport Psychol 2000; 31: 256-277.
- Singh MA. Exercise comes of age: rationale and recommendations for a geriatric exercise prescription. J Gerontol A Biol Sci Med Sci 2002; 57: 262-282.
- Onodera K, Sakurada S, Furuta S, et al. Age-related differences in forced walking stress-induced analgesia in mice. Drugs Exp Clin Res 2001; 27: 193-198.
- Koltyn KF, Trine MR, Stegner AJ, Tobar DA. Effect of isometric exercise on pain perception and blood pressure in men and women. Med Sci Sports Exerc 2001; 33: 282-290.
- Hoeger Bement MK, DiCapo J, Rasiarmos R, Hunter SK. Dose response of isometric contractions on pain perception in healthy adults. Med Sci Sports Exerc 2008; 40: 1880-1889.
- Umeda M, Newcomb LW, Ellingson LD, Koltyn KF. Examination of the dose-response relationship between pain perception and blood pressure elevations induced by isometric exercise in men and women. Biol Psychol 2010; 85; 90-96.
- Sternberg WF, Bokat C, Kass L, Alboyadjian A, Gracely RH. Sex dependent components of the analgesia produced by athletic competition. J Pain 2001; 2: 65-74.
- Hughes J, Smith T, Kosterlitz H, Fothergill LA, Morgan BA, Morris HR. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature 1975; 258: 577-580.
- Simantov R, Snyder S. Morphine-like peptides in a mammalian brain: isolation, structure elucidation, and interactions with an opiate receptor. Proc Natl Acad Sci USA 1976; 73: 2515-2519.
- Snyder SH, Pasternak GW. Historical review: opioid receptors. Trends Pharmacol Sci 2003; 24: 198-205.
- Connor M, Christie MD. Opioid receptor signalling mechanisms. Clin Exp Pharmacol Physiol 1999; 26: 493-499.
- Law PY, Wong YH, Loh HH. Molecular mechanisms and regulation of opioid receptor signaling. Annu Rev Pharmacol Toxicol 2000; 40: 389-430.
- Basbaum AI, Fields HL. The origin of descending pathways in the dorsolateral funiculus of the cord of the cat and rat: further studies on the anatomy of pain modulation. J Comp Neurol 1979; 187: 513-531.
- Willis WD, Haber LH, Martin RF. Inhibition of spinothalamic tract cells and intemeurons by brain stem stimulation in the monkey. J Neurophysiol 1977; 40: 968-981.
- Colt EW, Wardlaw SL, Frantz AG. The effect of running on plasma beta-endorphin. Life Sci 1981; 28: 1637-1640.
- Sforzo GA. Opioids and exercise. An update. Sports Med 1989; 7: 109-124.
- Howlett TA, Tomlin S, Ngahfoong L, et al. Release of beta endorphin and met-enkephalin during exercise in normal women: response to training. Br Med J 1984; 288: 1950-1952.
- Elliot DL, Goldberg L, Watts WJ, Orwoll E. Resistance exercise and plasma beta-endorphin/beta-lipotrophin immunoreactivity. Life Sci 1984; 34: 515-518.
- Kraemer WJ, Dziados JE, Marchitelli LJ, et al. Effects of different heavy-resistance exercise protocols on plasma beta-endorphin concentrations. J Appl Physiol 1993; 74: 450-459.
- Pierce EF, Eastman NW, Tripathi HT, Olson KG, Dewey WL. Plasma β-endorphin immunoreactivity: response to resistance exercise. J Sports Sci 1993; 11: 499-452.
- Pierce EF, Eastman NW, McGowan RW, Tripathi H, Dewey WL, Olson KG. Resistance exercise decreases beta-endorphin immunoreactivity. Br J Sports Med 1994; 28: 164-166.
- Olausson B, Eriksson E, Ellmarker L, Rydenhag B, Shyu BC, Andersson SA. Effects of naloxone on dental pulp pain threshold following muscle exercise and low frequency transcutaneous nerve stimulation: a comparative study in man. Acta Physiol Scand 1986; 126: 299-305.
- Droste C, Meyer-Blankenburg H, Greenlee MW, Roskamm H. Effect of physical exercise on pain thresholds and plasma beta-endorphins in patients with silent and symptomatic myocardial ischemia. Eur Heart J 1988; 9: 25-33.
- Janal MN, Colt EW, Clark WC, Glusman M. Pain sensitivity, mood and plasma endocrine levels in man following long distance running: effects of naloxone. Pain 1984; 19: 13-25.
- Haier RJ, Quaid K, Mills JS. Naloxone alters pain perception after jogging. Psychiatry Res 1981; 5: 231-232.
- Droste C, Greenlee MW, Schreck M, Roskamm H. Experimental pain thresholds and plasma beta-endorphin levels during exercise. Med Sci Sports Exerc 1991; 23: 334-342.
- Bodnar RJ, Kelly DD, Spiaggia A, Ehrenberg C, Glusman M. Dose-dependent reductions by naloxone of analgesia induced by cold-water stress. Pharmacol Biochem Behav 1978; 8: 667-672.
- O’Connor P, Chipkin RE. Comparisons between warm and cold-water stress in mice. Life Sci 1984; 35: 631-639.
- Willow M, Carmody J, Carroll P. The effects of swimming in mice on pain perception and sleeping time in response to hypnotic drugs. Life Sci 1980; 26: 219-224.
- Tierney G, Carmody J, Jamieson D. Stress analgesia: the opioid analgesia of long swims surpasses the non-opioid analgesia induced by short swims in mice. Pain 1991; 46: 89-95.
- Galdino GS, Duarte ID, Perez AC. Participation of endogenous opioids in the antinociception induced by resistance exercise in rats. Braz J Med Biol Res 2010; 43: 906-907.
- Koltyn KF. Using physical activity to manage pain in older adults. J Aging Phys Act 2002; 10: 226-239.
- Koltyn KF, Umeda M. Contralateral attenuation of pain after short-duration submaximal isometric exercise. J Pain 2007; 8: 887-892.
- Staud R, Robinson ME, Price DD. Isometric exercise has opposite effects on central pain mechanisms in fibromyalgia patients compared to normal controls. Pain 2005; 118: 176-184.
- Melchionda AM, Clarkson PM, Denko C, Freedson P, Graves J, Katch F. The Effect of local isometric exercise on serum levels of beta-endorphin/beta-lipotropin. Phys Sports Med 1984; 12: 102-109.
- Christie MJ, Chesher GB, Bird KD. The correlation between swim-stress induced antinociception and [3H] leu-enkephalin binding to brain homogenates in mice. Pharmacol Biochem Behav 1981; 15: 853-857.
- Shyu BC, Andersson SA, Thoren P. Endorphin mediated increase in pain threshold induced by long-lasting exercise in rats. Life Sci 1982; 30: 833-840.
- Bement MK, Sluka KA. Low-intensity exercise reverses chronic muscle pain in the rat in a naloxone-dependent manner. Arch Phys Med Rehabil 2005; 86: 1736-1740.
- Stagg NJ, Mata HP, Ibrahim MM, et al. Regular exercise reverses sensory hypersensitivity in a rat neuropathic pain model: role of endogenous opioids. Anesthesiology 2011; 114: 940-948.
- Bertolini GR, Rosa CT, Silva LI, Meireles A, Rocha BP. Use of resistance exercise as a factor antagonized by naloxone of analgesia in acute knee synovitis in Wistar rats. Rev Bras Med Esporte 2012; 18: 126-129.
- Hua S, Cabot PJ. Mechanisms of peripheral immune-cell-mediated analgesia in inflammation: clinical and therapeutic implications. Trends Pharmacol Sci 2010; 31: 427-433.
- Mazzardo-Martins L, Martins DF, Marcon R, et al. High-intensity extended swimming exercise reduces pain-related behavior in mice: involvement of endogenous opioids and the serotonergic system. J Pain 2010; 11: 1384-1393.
- Duarte ID, Nakamura M, Ferreira SH. Peripheral analgesia and activation of the nitric oxide cyclic GMP pathway. Eur J Pharmacol 1990; 186: 289-293.
- Jonsdottir IH, Jungersten L, Johansson C, Wennmalm A, Thorean P, Hoffmann P. Increase in nitric oxide formation after chronic voluntary exercise in spontaneously hypertensive rats. Acta Physiol Scand 1998; 162: 149-153.
- Dominiczak AF, Bohr DF. Nitric oxide and its putative role in hypertension. Hypertension 1995; 25: 1202-1211.
- Murad F. Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development. N Engl J Med 2006; 355: 2003-2011.
- Kawano T, Zoga V, Kimura M, et al. Nitric oxide activates ATP-sensitive potassium channels in mammalian sensory neurons: action by direct S-nitrosylation. Mol Pain 2009; 5: 12. doi: 10.1186/1744-8069-5-12
- McConell GK, Bradley SJ, Stephens TJ, Canny BJ, Kingwell BA, Lee-Young RS. Skeletal muscle nNOS mu protein content is increased by exercise training in humans. Am J Physiol Regul Integr Comp Physiol 2007; 293: R821-R828.
- Tschakovsky ME, Joyner MJ. Nitric oxide and muscle blood flow in exercise. Appl Physiol Nutr Met 2008; 33: 151-161.
- Reid M. Role of nitric oxide in skeletal muscle: synthesis, distribution and functional importance. Acta Physiol Scand 1998; 162: 401-409.
- Narin SO, Pinar L, Erbas D, Ozturk V, Idiman F. The effects of exercise and exercise-related changes in blood nitric oxide level on migraine headache. Clin Rehabil 2003; 17: 624-630.
- Galdino GS, Cortes SF, Duarte ID, Perez AC. Involvement of the nitric oxide/(C)GMP/K(ATP) pathway in antinociception induced by exercise in rats. Life Sci 2010; 86: 505-509.
- Galdino GS, Duarte ID, Perez AC. Central release of nitric oxide mediates antinociception induced by aerobic exercise. Braz J Med Biol Res 2015; 48: 790-797.
- Lovick TA. Role of nitric oxide in medullary raphe-evoked inhibition of neuronal activity in the periaqueductal gray matter. Neuroscience 1996; 75: 1203-1209.
- Spinella M, Bodnar RJ. Nitric oxide synthase inhibition selectively potentiates swim stress antinociception in rats. Pharmacol Biochem Behav 1994; 47: 727-733.
- Miller MJ, Thompson JH, Liu X, et al. Failure of L-NAME to cause inhibition of nitric oxide synthesis: role of inducible nitric oxide synthase. Inflamm Res 1996; 45: 272-276.
- Duarte ID, Ferreira SH. L-NAME causes antinociception by stimulation of the arginine-NO-cGMP pathway. Mediators Inflamm 2000; 9: 25-30.
- Cury Y, Picolo G, Gutierrez VP, Ferreira SH. Pain and analgesia: the dual effect of nitric oxide in the nociceptive system. Nitric Oxide 2011; 25: 243-254.
- Kawabata A, Manabe S, Manabe Y, Takagi H. Effect of topical administration of L-arginine on formalin-induced nociception in the mouse: a dual role of peripherally formed NO in pain modulation. Br J Pharmacol 1994; 112: 547-550.
- Sousa AM, Prado WA. The dual effect of a nitric oxide donor in nociception. Brain Res 2001; 897: 9-19.
- Rocha J, Peixoto ME, Jancar S, Cunha FQ, Ribero RA, da Rocha FA. Dual effect of nitric oxide in articular inflammatory pain in zymosan-induced arthritis in rats. Br J Pharmacol 2002; 136: 588-596.
- Rapport MM, Green AA, Page IH. Serum vasoconstrictor, serotonin; isolation and characterization. J Biol Chem 1948; 176: 1243-1251.
- Hoyer D, Hannon JP, Martin GR. Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol Biochem Behav 2002; 71: 533-554.
- Millan MJ. Descending control of pain. Prog Neurobiol 2002; 66: 355-474.
- Rivot JP, Weil-Fugazza J, Godefroy F, Bineauthurotte M, Orylavollee L, Besson JM. Involvement of serotonin in both morphine and stimulation-produced analgesia: electrochemical and biochemical approaches. Adv Pain Res Ther 1984; 6: 135-150.
- Hammond DL, Tyce GM, Yaksh TL. Efflux of 5-hydroxytryptamine and noradrenaline into spinal cord superfusates during stimulation of the rat medulla. J Physiol 1985; 359: 151-162.
- Liang DY, Li X, Clark JD. 5-hydroxytryptamine type 3 receptor modulates opioid-induced hyperalgesia and tolerance in mice. Anesthesiology 2011; 114: 1180-1189.
- Chu LF, Liang DY, Li X, et al. From mouse to man: the 5-HT3 receptor modulates physical dependence on opioid narcotics. Pharmacogenet Genomics 2009; 19: 193-205.
- Kiefel JM, Paul D, Bodnar RJ. Reduction in opioid and nonopioid forms of swim analgesia by 5HT-2 antagonists. Brain Res 1989; 500: 231-240.
- Brown BS, Payne T, Kim C, Moore G, Krebs P, Martin W. Chronic response of rat brain norepinephrine and serotonin levels to endurance training. J Appl Physiol Respir Environ Exerc Physiol 1979; 46: 19-23.
- Gerin C, Teilhac JR, Smith K, Privat A. Motor activity induces release of serotonin in the dorsal horn of the rat lumbar spinal cord. Neurosci Lett 2008; 436: 91-95.
- Korb A, Bonetti LV, da Silva SA, et al. Effect of treadmill exercise on serotonin immunoreactivity in medullary raphe nuclei and spinal cord following sciatic nerve transection in rats. Neurochem Res 2010; 35: 380-389.
- Sokunbi O, Watt P, Moore A. Changes in plasma concentration of serotonin in response to spinal stabilisation exercises in chronic low back pain patient. Nig Q J Hosp Med 2007; 17: 108-111.
- Sokunbi O, Moore A, Watt P. Plasma levels of beta-endorphin and serotonin in response to specific spinal based exercises. S Afr J Physiother 2008; 64: 31-37.
- Valim V, Natour J, Xiao Y, et al. Effects of physical exercise on serum levels of serotonin and its metabolite in fibromyalgia: a randomized pilot study. Rev Bras Reumatol 2013; 53: 538-541.
- Lundberg A, Vyklicky L. Inhibitory interaction between spinal reflexes to primary afferents. Experientia 1963; 19: 247-248.
- Yaksh TL. Pharmacology of spinal adrenergic systems which modulate spinal nociceptive processing. Pharmacol Biochem Behav 1985; 22: 845-858.
- MacDonald E, Kobilka BK, Scheinin M. Gene targeting-homing in on alpha 2-adrenoceptor-subtype function. Trends Pharmacol Sci 1997; 18: 211-219.
- Nicholas AP, Hokfelt T, Pieribone VA. The distribution and significance of CNS adrenoceptors examined with in situ hybridization. Trends Pharmacol Sci 1996; 17: 245-255.
- Bodnar RJ, Mann PE, Stone EA. Potentiation of cold-water swim analgesia by acute, but not chronic desipramine administration. Pharmacol Biochem Behav 1985; 23: 749-752.
- Oluyomi AO, Hart SL. Alpha-adrenoceptor involvement in swim stress-induced antinociception in the mouse. J Pharm Pharmacol 1990; 42: 778-784.
- Tokuyama S, Takahashi M, Kaneto H. Participation of an alpha 2-mediated mechanism in the production of forced swimming-stress induced analgesia in mice. J Pharmacobiodyn 1991; 14: 357-361.
- Tokuyama S, Takahashi M, Kaneto H. Further evidence for the participation of an alpha 2-adrenoceptor mediated mechanism in the production of forced swimming-stress induced analgesia in mice. J Pharmacobiodyn 1991; 14: 637-641.
- de Souza GG, Duarte ID, de Castro Perez A. Differential involvement of central and peripheral a2 adrenoreceptors in the antinociception induced by aerobic and resistance exercise. Anesth Analg 2013; 116: 703-711.
- Pertwee RG. Cannabinoid receptors and pain. Prog Neurobiol 2001; 63: 569-611.
- Fan Y, Hooker BA, Garrison, TR, et al. Pharmacological and molecular characterization of a dorsal root ganglion cell line expressing cannabinoid CB (1) and CB (2) receptors. Eur J Pharmacol 2011; 659: 161-168.
- Di Marzo V, Bifulco M, De Petrocellis L. The endocannabinoid system and its therapeutic exploitation. Nat Rev Drug Discov 2004; 3: 771-784.
- Sparling PB, Giuffrida A, Piomelli D, Rosskopf L, Dietrich A. Exercise activates the endocannabinoid system. Neuroreport 2003; 14: 2209-2211.
- Onaivi ES, Carpio O, Ishiguro H, Schanz N, Uhl GR, Benno R. Behavioral effects of CB2 cannabinoid receptor activation and its influence on food and alcohol consumption. Ann NY Acad Sci 2008; 1139: 426-433.
- Rawls SM, Benamar K. Effects of opioids, cannabinoids, and vanilloids on body temperature. Front Biosci 2011; 3: 822-845.
- Keeney BK, Raichlen DA, Meek TH, et al. Differential response to a selective cannabinoid receptor antagonist (SR141716: rimonabant) in female mice from lines selectively bred for high voluntary wheel-running behaviour. Behav Pharmacol 2008; 19: 812-820.
- Heyman E, Gamelin FX, Goekint M, et al. Intense exercise increases circulating endocannabinoid and BDNF levels in humans - possible implications for reward and depression. Psychoneuroendocrinology 2012; 37: 844-851.
- Raichlen DA, Foster AD, Seillier A, Giuffrida A, Gerdeman GL. Exercise-induced endocannabinoid signaling is modulated by intensity. Eur J Appl Physiol 2013; 13: 869-875.
- Hill MN, Titterness AK, Morrish AC, et al. Endogenous cannabinoid signaling is required for voluntary exercise-induced enhancement of progenitor cell proliferation in the hippocampus. Hippocampus 2010; 20: 513-523.
- Koltyn KF, Brellenthin AG, Cook DB, Sehgal N, Hillard C. Mechanisms of exercise-induced hypoalgesia. J Pain 2014; 15: 1294-1304.
- Crombie KM, Brellenthin AG, Hillard CJ, Koltyn KF. Endocannabinoid and opioid system interactions in exercise-induced hypoalgesia. Pain Med 2017; 19: 118-123.
- Galdino G, Romero T, Silva JF, et al. Acute resistance exercise induces antinociception by activation of the endocannabinoid system in rats. Anesth Analg 2014; 119: 702-715.
- Galdino G, Romero TR, Silva JF, et al. The endocannabinoid system mediates aerobic exercise-induced antinociception in rats. Neuropharmacology 2014; 77: 313-324.
- Fuss J, Steinle J, Bindila L, et al. A runner’s high depends on cannabinoid receptors in mice. Proc Natl Acad Sci USA 2015; 112: 13105-13108.
- King-Himmelreich TS, Moser CV, Wolters MC, et al. AMPK contributes to aerobic exercise-induced antinociception downstream of endocannabinoids. Neuropharmacology 2017; 24: 134-142.
- Mota BC, Pereira L, Souza MA, et al. Exercise pre-conditioning reduces brain inflammation and protects against toxicity induced by traumatic brain injury: behavioral and neurochemical approach. Neurotox Res 2012; 21: 175-184.
- Leung A, Gregory NS, Allen LA, Sluka KA. Regular physical activity prevents chronic pain by altering resident muscle macrophage phenotype and increasing interleukin-10 in mice. Pain 2016; 157: 70-79.
- Kawanishi N, Yano H, Yokogawa Y, Suzuki K. Exercise training inhibits inflammation in adipose tissue via both suppression of macrophage infiltration and acceleration of phenotypic switching from M1 to M2 macrophages in high-fat-diet-induced obese mice. Exerc Immunol Rev 2010; 16: 105-118.
- Bobinski F, Teixeira JM, Sluka KA, Santos AR. Interleukin-4 mediates the analgesia produced by low-intensity exercise in mice with neuropathic pain. Pain 2018; 159: 437-450.
- Watkins LR, Maier SF. Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov 2003; 12: 973-985.
- Jung JS, Shin JA, Park EM, Lee JE, Kang YS. Anti-inflammatory mechanism of ginsenoside Rh1 in lipopolysaccharide-stimulated microglia: critical role of the protein kinase A pathway and hemeoxygenase-1 expression. J Neurochem 2010; 6: 1668-1680.
- Watkins LR, Milligan ED, Maier SF. Glial proinflammatory cytokines mediate exaggerated pain states: implications for clinical pain. Adv Exp Med Biol 2003; 521: 1-21.
- Gong X, Chen Y, Fu B, Jiang J, Zhang M. Infant nerve injury induces delayed microglial polarization to the M1 phenotype, and exercise reduces delayed neuropathic pain by modulating microglial activity. Neuroscience 2017; 349: 76-86.
- Sakr HF, Abbas AM, El Samanoudy AZ. Effect of vitamin E on cerebral cortical oxidative stress and brain-derived neurotrophic factor gene expression induced by hypoxia and exercise in rats. J Physiol Pharmacol 2015; 66: 191-202.
- Verbickas V, Baranauskiene N, Eimantas N, et al. Effect of sprint cycling and stretch-shortening cycle exercises on the neuromuscular, immune and stress indicators in young men. J Physiol Pharmacol 2017; 68: 125-132.
- Siuciak JA, Altar CA, Wiegand SJ, Lindsay RM. Antinociceptive effect of brain-derived neurotrophic factor and neurotrophin-3. Brain Res 1994; 633: 326-330.
- Siuciak JA, Wong V, Pearsall D, Wiegand SJ, Lindsay RM. BDNF produces analgesia in the formalin test and modifies neuropeptide levels in rat brain and spinal cord areas associated with nociception. Eur J Neurosci 1995; 7: 663-670.
- Bostrom P, Wu J, Jedrychowski MP, et al. A PGC1-alphadependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012; 481: 463-468.
- Wrann CD, White JP, Salogiannnis J, et al. Exercise induces hippocampal BDNF through a PGC-1alpha/FNDC5 pathway. Cell Metab 2013; 18: 649-659.
- Hofmann T, Elbelt U, Ahnis A, et al. The exercise-induced myokine irisin does not show an association with depressiveness, anxiety and perceived stress in obese women. J Physiol Pharmacol 2016; 67: 195-203.
- Pacheco DF, Reis GML, Francischi JN, Castro MSA, Perez AC, Duarte IDG. d-opioid receptor agonist SNC80 elicits peripheral antinociception via d1 and d2 receptors and activation of the L-arginine/nitric oxide/cyclic GMP pathway. Life Sci 2005; 78: 54-60.
- Hamalainen MM, Lovick TA. Involvement of nitric oxide and serotonin in modulation of antinociception and pressor responses evoked by stimulation in the dorsolateral region of the periaqueductal gray matter in the rat. Neuroscience 1997; 80: 821-827.
- Reis GM, Pacheco D, Perez AC, Klein A, Ramos MA, Duarte ID. Opioid receptor and NO/cGMP pathway as a mechanism of peripheral antinociceptive action of the cannabinoid receptor agonist anandamide. Life Sci 2009; 85: 351-356.
- Negrete R, Hervera A, Leanez S, Martin-Campos JM, Pol O. The antinociceptive effects of JWH-015 in chronic inflammatory pain are produced by nitric oxide-cGMP-PKG-KATP pathway activation mediated by opioids. PLoS One 2011; 6: e26688. doi: 10.1371/journal.pone.0026688
- Romero TR, Duarte ID. Alpha (2)-Adrenoceptor agonist xylazine induces peripheral antinociceptive effect by activation of the L-arginine/nitric oxide/cyclic GMP pathway in rat. Eur J Pharmacol 2009; 613: 64-67.
- Lichtman AH, Martin BR. Cannabinoid-induced antinociception is mediated by a spinal alpha 2-noradrenergic mechanism. Brain Res 1991; 559: 309-314.
A c c e p t e d : January 30, 2018